CS 5043: HW5: Recurrent Neural Networks and String Data
Assignment notes:
- Deadline: Thursday, April 7th @11:59pm.
- Hand-in procedure: submit a zip file to Gradescope
- This work is to be done on your own. While general discussion
about Python, Keras and Tensorflow is encouraged, sharing
solution-specific code is inappropriate. Likewise, downloading
solution-specific code is not allowed.
- Do not submit MSWord documents.
The Problem
Peptides are short chains of amino acids that perform a wide range of
biological functions, depending on the specific sequence of amino
acids. Understanding how amino acids interact with other molecules is
key to understanding the mechanisms of genetic diseases and to
constructing therapies to address these diseases.
One form of interaction is the question of how well a peptide binds to
a specific molecule (this is known as binding affinity). For
this assignment, we will use a data set that includes a measure of
binding affinity for a large number of peptides toward two molecules,
known as HLA-DRB1*13:01 and HLA-DRB1*15:01. Our goal is to predict
the binding affinity for novel peptides toward these molecules.
Amino Acids
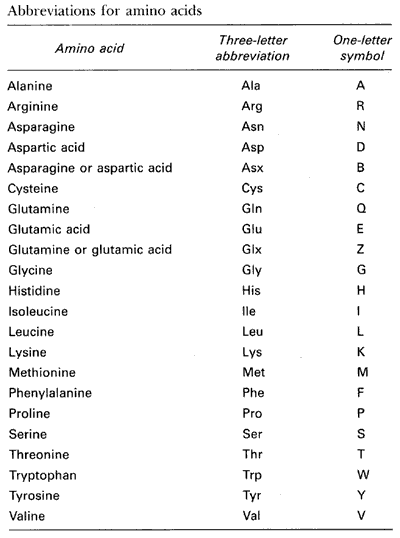
A total of 22 amino acids make up the building blocks for all peptides
and proteins. The following table gives the names and one-letter
codes used to represent these amino acids:
Data Set
The Data set is available on SCHOONER:
- /home/fagg/datasets/hlas: directory tree containing the data
- /home/fagg/datasets/HLA.zip: zip file containing all of the data
The data are already partitioned into five independent folds
of training and validation data. For the purposes of this assignment,
you should preserve this partition. Also, you should only use the
HLA-DRB1*13:01 molecule (look for files containing "1301").
Each row in the CSV files contains two columns: a string representing
the sequence of amino acids and a measure of binding affinity. Each
character of the string corresponds to a unique amino acid. Here
is an example:
DLDKKETVWHLEE,0
DKSKPKVYQWFDL,0
HYTVDKSKPKVYQ,0
KSKPKVYQWFDLR,0
LHYTVDKSKPKVY,0
TVDKSKPKVYQWF,0
VDKSKPKVYQWFD,0
YTVDKSKPKVYQW,0
ADVILPIGTRSVETD,0.646144
AFKPVLVDEGRKVAI,0.57075
AGLKTNDRKWCFEGP,0.615622
AHLAEENEGDNACKR,0
ALYEKKLALYLLLAL,0.610019
ANGKTLGEVWKRELN,0.495407
Note that different rows contain different numbers of amino acids.
The binding affinity is encoded on a log scale and ranges between 0
and 1. Peptides with affinities at or above 0.426 are considered
more likely than not to bind to the molecule in question, whereas
affinities below this threshold are considered less likely to bind.
Deep Learning Experiment
Objective: Create a recurrent neural network that predicts binding
as a function of the amino acid string.
- The provided prepare_data_set() function (described below) will
load the specified rotation of the data. The amino acid
strings are already converted to a RNN-ready format (examples x
time: int)
- Use an embedding layer to translate the individual ints to
a k-dimensional space
- Follow the embedding layer with one or more trainable layer(s).
- Use your favorite RNN module (use return_sequences=False)
- Use one or more dense layers to predict either the affinity
measure (a continuous problem) or whether the peptide will
bind to the molecule (a binary problem).
Think about what the appropriate non-linearity is for the
output unit
Performance Reporting
Once you have selected a reasonable architecture and set of
hyper-parameters, produce the following figures:
- Figure 1: Training set Area Under the Curve (AUC) as a function of epoch for each of
the rotations (so, 5 curves)
- If you are solving the binary prediction problem, use
tf.keras.metrics.AUC() and set the threshold
to 0.426
- If you are solving the continous prediction problem, use
MyAUC()
- Figure 2: Validation set AUC as a function of epoch for each of the rotations
- Figure 3: Training set Accuracy as a function of epoch for each
rotation.
- If you are solving the binary prediction problem, use
tf.keras.metrics.BinaryAccuracy() and set the threshold
to 0.426
- If you are solving the continous prediction problem, use
MyBinaryAccuracy()
- Figure 4: Validation set accuracy as a function of epoch for each of the
rotations
- Figure 5: Histogram of AUC for the test folds that shows a
vertical line that corresponds to the average AUC (also show
this average in text). An average test AUC of 0.85 is doing well
- Figure 6: Histogram of accuracy for the test folds that shows a
vertical line that corresponds to the average accuracy (also
show this average in text).
Provided Code
In code for class:
- hla_support.py: prepare_data_set() will load and prepare a data set for training/validation/testing
- The "ins" are token indices (not a 1-hot encoding).
- This function splits the training data set into a
training and validation data set. Leave the seed and
the size at their default values (this way, we can
better compare our results)
- Hyper-parameter searches should only be done using the
performance of the validation data set
- You can evaluate the performance of the model on the
test set using:
model.evaluate(ins_test, outs_test)
BUT: only look at these values after you have selected your favorite hyper-parameters
- Use
a tf.keras.layers.Embedding as the first layer of
your network - it will properly handle the token indices
- metrics_binarized.py: Useful metrics
- The "outs" from the data loader are log affinity measures. However, any
peptide with an affinity at or above 0.426 is considered
as "bindable" to the molecule in question ("bindable" is a
binary question).
- You may formulate your problem entirely in terms of a
binary prediction problem, in which case you will
convert the "outs" to binary vectors and use
cross-entropy as your loss. This conversion happens
before you begin training
- You may also choose to predict the log affinity
directly, in which case you might use mse as your loss.
However, in order to report Accuracy or AUC, you will
need to binarize the true values of the outs. The
provided wrapper classes do just this for you.
- MyBinaryAccuracy: metric class that takes as
input continuous values for both the true values
and predicted ones. The constructor for this
class takes as input a threshold that is used to
determine whether the continuous values are in
the "true" or "false" class. Use the binding
threshold for this threshold.
- MyAUC: metric class that takes as input
continuous values for both th true values and
predicted ones. The constructor also takes as
input a threshold that is used to determine
whether the continuous value for the true value
corresponds to the "true" or "false" class. Use the binding
threshold for this threshold.
Remember that AUC then imagines all possible
thresholds for the predicted value. For each of
these thresholds, it computes both the true
positive rate and the false positive rate,
yielding our ROC curve. AUC is the integral of
this curve over the 0 ... 1 range.
What to Hand In
Turn in a single zip file that contains:
- All of your python code (.py) and any notebook file (.ipynb)
[Gradescope can render notebook files directly - no need to
convert to pdf!]
- A picture of your model architecture (from plot_model())
- Figures 1-6
Grading
- 20 pts: Clean code for model building (including in-code documentation)
- 10 pts: Figure 1
- 10 pts: Figure 2
- 10 pts: Figure 3
- 10 pts: Figure 4
- 10 pts: Figure 5
- 10 pts: Figure 6
- 20 pts: Reasonable test set performance for all rotations
References
andrewhfagg -- gmail.com
Last modified: Fri Apr 1 20:37:47 2022